
La maladie de Creutzfeldt-Jakob est une maladie neurodégénérative rare qui affecte rapidement, progressivement et sévèrement le cerveau. Ce trouble, bien que rare, suscite une attention croissante en raison de son impact dévastateur sur les patients et leurs familles.
La maladie de Creutzfeldt-Jakob (MCJ) détruit progressivement les cellules du cerveau et cause de minuscules trous dans ce dernier. Les personnes atteintes de la MCJ souffrent d’ataxie ou ont de la difficulté à contrôler les mouvements du corps, ce qui entraîne une démarche anormale, des troubles de la parole et une démence. C’est toujours fatal, et il n’existe actuellement aucun remède.
Chaque année, la MCJ touche une personne sur un million dans le monde, y compris aux États-Unis. Les causes de cette maladie peuvent être sporadiques, héréditaires ou acquises. Elle affecte principalement les personnes de plus de 60 ans, et il est rare de la diagnostiquer chez des individus de moins de 30 ans.
Il est peu probable qu’une personne atteinte de MCJ survive plus d’un an après l’apparition des symptômes, ce qui souligne l’urgence d’une prise en charge médicale adéquate.
Qu’est-ce que la MCJ?

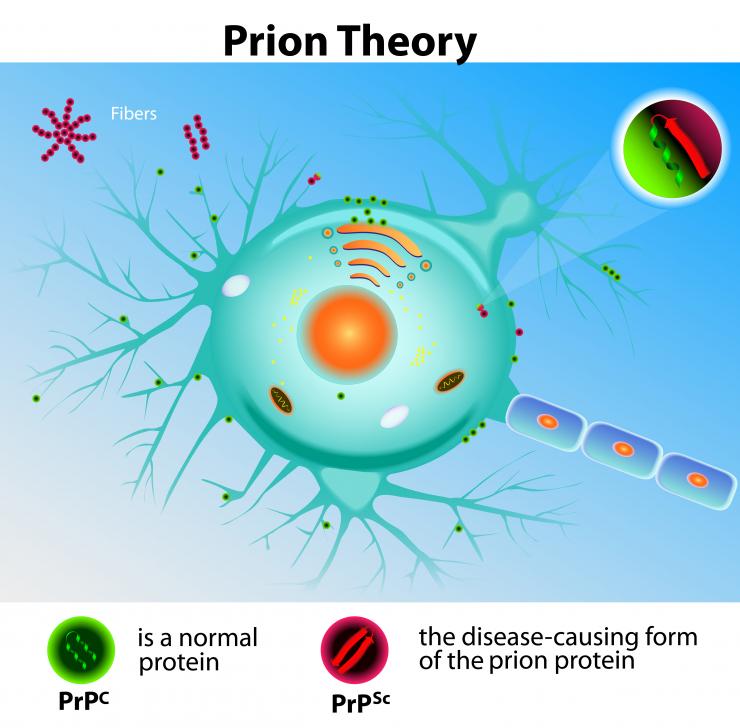
La MCJ est une encéphalopathie spongiforme transmissible (EST) qui détruit le cerveau au fil du temps, causée par un agent infectieux connu sous le nom de prion. Contrairement aux virus ou aux bactéries, les prions sont des protéines mal repliées qui induisent des modifications structurelles dans d’autres protéines cérébrales.
D’autres types d’EST incluent le syndrome de Gerstmann-Sträussler-Scheinker (GSS), l’insomnie familiale fatale et le kuru chez l’homme. Des exemples chez les animaux incluent la tremblante du mouton et de la chèvre, ainsi que l’encéphalopathie spongiforme bovine (ESB), également connue sous le nom de « maladie de la vache folle ».
Les Centers for Disease Control and Prevention (CDC) notent que la MCJ classique n’est pas liée à l’ESB ou à d’autres variantes, bien que des recherches soient en cours pour mieux comprendre ces liens.
Symptômes
La MCJ présente une longue période d’incubation, avec des symptômes pouvant prendre jusqu’à 40 ans avant d’apparaître. Ces symptômes surviennent lorsque les cellules cérébrales sont détruites, entraînant une détérioration rapide de l’état du patient en quelques semaines, la plupart des patients décédant dans l’année qui suit.
Les symptômes caractéristiques de la MCJ incluent une progression rapide vers la démence et des myoclonies, qui sont des mouvements involontaires spasmodiques des muscles. D’autres symptômes peuvent comprendre des changements d’humeur, des pertes de mémoire et un jugement altéré.
Bien que la MCJ puisse ressembler à la maladie d’Alzheimer ou à la maladie de Huntington, les symptômes évoluent beaucoup plus rapidement, en quelques jours ou semaines. À mesure que la maladie progresse, des problèmes de coordination et de vision apparaissent, pouvant conduire à la cécité. En fin de maladie, le patient perd la capacité de bouger ou de parler et entre dans un coma.
Des études autopsiques de tissu cérébral ont montré que la MCJ présente des changements uniques qui ne se retrouvent pas dans d’autres types de démence.
Il existe plusieurs variantes de la MCJ qui peuvent ne pas être directement liées à la forme classique et dont les symptômes ainsi que l’évolution peuvent diverger.
Causes
La MCJ se produit lorsque des prions, des protéines anormales, provoquent des anomalies dans d’autres protéines cérébrales. Cette accumulation et malformation des prions sur les cellules cérébrales entraînent des lésions cérébrales et la mort cellulaire.
La maladie peut être sporadique, héréditaire ou acquise.
CJD sporadique
Dans 85% des cas, la MCJ est sporadique, sans facteurs de risque apparents.
CJD héritée
Entre 5% et 10% des cas sont héréditaires, résultant de mutations dans le gène associé à la formation des prions. Des antécédents familiaux de MCJ peuvent être présents, ou une mutation peut se produire dans les ovules ou spermatozoïdes, exposant ainsi la descendance à un risque accru de développer la maladie.
Les prions, ne contenant pas d’information génétique, ne nécessitent pas de gènes pour se reproduire, mais une mutation dans le gène de la protéine prion normale peut provoquer des anomalies des prions. Plusieurs mutations spécifiques du gène prion ont été identifiées, chacune influençant la fréquence d’apparition de la maladie et les symptômes observés.
Cependant, toutes les personnes porteuses de mutations dans le gène des prions ne développent pas nécessairement la MCJ.
CJD acquise
Bien qu’aucune preuve n’indique que la MCJ peut se transmettre d’une personne à une autre, certaines procédures médicales ont été associées à la transmission de la maladie. Ces procédures comprennent :
- greffe de cornée
- implants d’électrode
- greffe de dure-mère, qui est le tissu qui recouvre le cerveau
- utilisation d’hormone de croissance humaine
Environ 1% des cas sont transmis par une exposition connue ou suspectée à des tissus cérébraux ou nerveux infectés.
Encéphalopathie spongiforme bovine
Dans les années 1990, un type de MCJ a été lié à l’exposition à l’ESB, une maladie touchant les bovins. On pensait que cette transmission était liée à la consommation de produits alimentaires contaminés. Cette variante a généralement affecté des patients plus jeunes et a eu une évolution plus prolongée.
L’ESB touche plusieurs espèces, y compris les bovins, les humains et même les chats. Bien que certains scientifiques pensent qu’un « virus lent » ou un autre organisme pourrait être à l’origine de la MCJ, aucun agent spécifique n’a encore été isolé chez les personnes atteintes de cette maladie.
L’agent responsable de la MCJ présente des caractéristiques atypiques pour un virus ou une bactérie, comme une longue période d’incubation, une résistance à la destruction et l’absence d’informations génétiques sous forme d’acides nucléiques.
Ainsi, les scientifiques concluent que la MCJ et d’autres EST sont causées non par un organisme vivant, mais par des prions, qui sont des protéines mal repliées provoquant des dommages au tissu cérébral et entraînant les symptômes caractéristiques de la MCJ.
Diagnostic
Il n’existe pas de test spécifique pour confirmer le diagnostic de MCJ. Seule une biopsie cérébrale pourrait le faire, mais cela représente un risque trop élevé pour le patient vivant.
Cependant, divers tests peuvent aider à identifier la cause probable des symptômes. Un examen physique peut mettre en évidence des spasmes musculaires et vérifier les réflexes du patient, qui peuvent être anormalement réactifs. Les muscles peuvent également présenter une tonicité excessive ou une flaccidité, selon la partie du cerveau affectée par la maladie.
Un examen de la vue peut révéler une cécité partielle non détectée par le patient. Un électroencéphalogramme (EEG) peut montrer des impulsions électriques anormales, tandis qu’une tomodensitométrie ou une IRM peut permettre d’exclure un accident vasculaire cérébral comme cause des symptômes.
Une ponction lombaire pour analyser le liquide céphalo-rachidien peut également être pratiquée, afin d’éliminer d’autres causes de démence. Si la protéine 14-3-3 est détectée dans le liquide et que le patient présente des symptômes typiques, il existe de fortes chances que la personne ait la MCJ.
Les biopsies cérébrales post-mortem montrent des tissus cérébraux spongieux, avec de minuscules trous visibles là où des amas de cellules nerveuses ont été détruits.
Traitement
Actuellement, il n’existe aucun remède contre la MCJ, et aucun médicament n’est capable de ralentir la progression de la maladie. Le traitement vise donc à soulager les symptômes et à rendre le patient aussi confortable que possible.
Les analgésiques opioïdes peuvent aider à soulager la douleur, tandis que des médicaments comme le clonazépam et le valproate de sodium peuvent atténuer les mouvements involontaires, tels que les contractions musculaires. Dans les stades avancés, il est souvent nécessaire de déplacer le patient fréquemment pour prévenir les escarres. Des méthodes telles que l’utilisation de cathéters pour drainer l’urine et l’alimentation par intraveineuse peuvent également être mises en place.
Prévention
Les mesures préventives incluent la stérilisation de tout équipement médical pour éliminer les agents pathogènes potentiels et le refus de dons de cornées provenant de personnes ayant un diagnostic de MCJ connu ou suspecté. La plupart des pays ont désormais des directives strictes concernant la gestion des bovins infectés et des restrictions sur les aliments pour animaux, réduisant ainsi le risque de transmission d’autres formes d’EST à l’homme.
Il est conseillé aux personnes ayant été exposées à des individus diagnostiqués avec la MCJ de suivre certaines recommandations, telles que :
- couvrir les plaies ouvertes, les coupures et les abrasions sur la peau
- porter des gants lors de la manipulation de tissus, du sang ou d’autres fluides corporels
- utiliser une blouse ou des vêtements jetables en cas de contact avec un patient
- employer un écran facial, des lunettes de protection ou un masque en cas de risque d’éclaboussement de fluides contaminés
- stériliser tout équipement utilisé sur ou à proximité du patient
Des recherches sont en cours sur le rôle des prions dans la MCJ, dans le but de mieux comprendre comment la maladie affecte le cerveau et de découvrir un traitement efficace. De nouvelles études s’attachent à explorer les mécanismes sous-jacents de cette maladie complexe, espérant ainsi un jour ouvrir la voie à des options thérapeutiques prometteuses.







