
Le syndrome d’Apert est une maladie génétique rare qui provoque la fusion prématurée des os du visage et du crâne d’un fœtus. Cette condition complexe peut avoir des conséquences significatives sur le développement physique et neurologique de l’enfant.
Les anomalies causées par le syndrome d’Apert peuvent engendrer des déficiences visuelles, des problèmes dentaires, ainsi que des malformations au niveau des doigts et des orteils, souvent accompagnées de syndactylie. Cet article propose une exploration approfondie du syndrome d’Apert, en examinant ses symptômes, ses traitements, et les perspectives actuelles sur cette maladie.
Qu’est-ce que le syndrome d’Apert?
Le syndrome d’Apert est une affection génétique où les os du crâne fusionnent trop tôt, affectant ainsi la forme de la tête et du visage. Les personnes atteintes peuvent faire face à diverses complications, notamment des problèmes de vision et des malocclusions dentaires dues à la déformation des structures faciales.
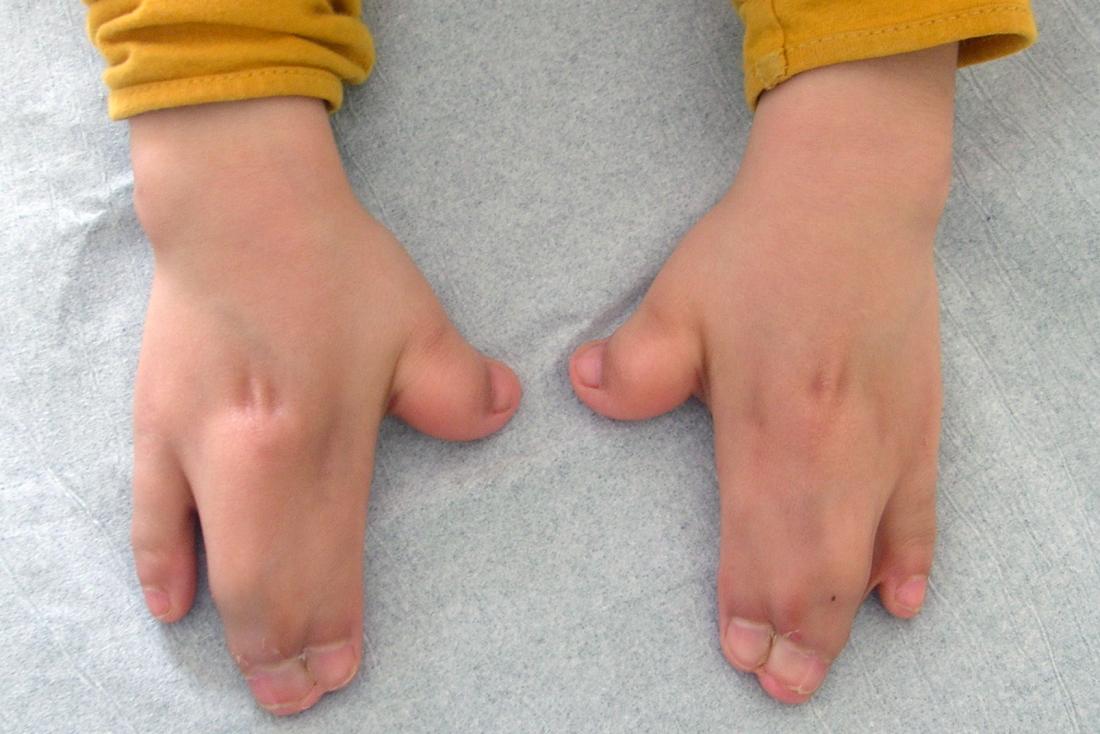
Dans de nombreux cas, trois doigts ou orteils fusionnent, ce qui est connu sous le nom de syndactylie. Les manifestations cliniques de ce syndrome sont variées, ce qui peut compliquer le diagnostic précoce.
Cette condition est généralement causée par une mutation génétique, se présentant parfois sans antécédents familiaux, mais pouvant également être héritée d’un parent. La compréhension de cette maladie continue d’évoluer avec la recherche médicale.
Symptômes
Les symptômes caractéristiques du syndrome d’Apert comprennent:
- un crâne en forme de cône, connu sous le nom de turribrachycéphalie
- un visage profondément enfoncé au milieu
- des yeux larges et bombés vers l’extérieur
- un nez becqué
- une mâchoire supérieure sous-développée, entraînant souvent un surpeuplement dentaire
Les anomalies faciales et crâniennes peuvent entraîner des problèmes de santé à long terme. Si elles ne sont pas corrigées, des problèmes de vision peuvent survenir en raison d’orbites peu profondes, et la petite taille de la mâchoire supérieure peut compliquer la dentition. De plus, la pression exercée sur le cerveau en développement peut affecter le développement cognitif, avec des individus atteints présentant un niveau intellectuel souvent moyen ou une déficience intellectuelle légère à modérée.
Les enfants nés avec le syndrome d’Apert présentent souvent des doigts et des orteils palmés. Bien que la fusion touche généralement trois doigts ou orteils, il arrive que l’ensemble d’un groupe de doigts soit fusionné. Plus rarement, des doigts ou orteils supplémentaires peuvent être présents.
D’autres signes et symptômes associés au syndrome d’Apert incluent:
- perte auditive
- acné sévère
- transpiration abondante
- fusion des os de la colonne vertébrale dans le cou
- peau grasse
- absence de poils dans les sourcils
- retards de croissance et de développement
- infections récurrentes de l’oreille
- fente palatine
- déficience intellectuelle légère à modérée
Causes

Le syndrome d’Apert est causé par une mutation d’un gène spécifique, souvent le gène FGFR2 ou FGFR1, responsable de la production d’une protéine essentielle dans le développement embryonnaire. Cette mutation peut survenir sporadiquement ou être héritée d’un parent, ce qui rend son apparition difficile à prédire.
La protéine résultante joue un rôle crucial dans la signalisation des cellules osseuses. Une mutation entraîne une signalisation excessive, provoquant ainsi la fusion prématurée des os du crâne, du visage, ainsi que des membres.
Les mutations du gène peuvent également être responsables d’autres troubles, tels que:
- le syndrome de Pfeiffer
- le syndrome de Crouzon
- le syndrome de Jackson-Weiss
Fréquence et héritage
Le syndrome d’Apert demeure une maladie extrêmement rare. Les estimations concernant sa prévalence varient considérablement. Selon la Bibliothèque nationale américaine de médecine, il affecte environ 1 nouveau-né sur 65 000 à 88 000, tandis que l’Organisation nationale pour les maladies rares (NORD) estime que ce chiffre serait plutôt de 1 sur 165 000 à 200 000 naissances.
La plupart des cas se présentent sans antécédents familiaux, mais lorsque l’un des parents est atteint, l’enfant a 50 % de chances de développer le syndrome à chaque grossesse. La répartition entre les sexes semble être équitable, touchant aussi bien les garçons que les filles.
Diagnostic
Le diagnostic du syndrome d’Apert peut souvent être établi à la naissance ou dans les premiers mois de la vie. Les médecins recherchent des anomalies osseuses typiques affectant la tête, le visage, les mains et les pieds pour confirmer le diagnostic.
Des examens d’imagerie comme des radiographies ou des tomodensitogrammes peuvent être réalisés pour évaluer les malformations osseuses. Les tests génétiques peuvent également apporter des éléments supplémentaires pour le diagnostic et la confirmation de la mutation génétique.
Traitement

Le traitement du syndrome d’Apert est personnalisé, s’adaptant aux besoins spécifiques de chaque patient. Les médecins collaborent étroitement avec les familles pour élaborer des plans de traitement adaptés à la gravité des symptômes.
Dans certains cas, une intervention chirurgicale peut être nécessaire pour réduire la pression sur le cerveau. D’autres procédures peuvent être envisagées, comme le remodelage des structures faciales ou la séparation des doigts ou orteils fusionnés.
Les enfants atteints du syndrome d’Apert nécessitent un suivi régulier tout au long de leur vie. Les médecins surveillent les complications liées à la maladie et proposent des traitements adaptés. Parmi les soins courants, on trouve:
- la correction des problèmes de vision
- des thérapies pour aider à surmonter les retards de développement
- des interventions dentaires pour remédier aux problèmes de surpeuplement dentaire
Perspective
Un diagnostic précoce est crucial pour améliorer les chances de traiter efficacement les symptômes et les complications associées au syndrome d’Apert. Bien que cela n’affecte généralement pas l’espérance de vie, des problèmes cardiaques ou d’autres complications peuvent survenir.
Les perspectives à long terme pour les personnes atteintes du syndrome d’Apert se sont améliorées grâce aux avancées de la médecine moderne. Les traitements peuvent souvent améliorer la qualité de vie et les résultats fonctionnels des patients.
Un enfant atteint du syndrome d’Apert devrait bénéficier d’un suivi médical continu, incluant des thérapies pour aider à faire face aux défis quotidiens que pose cette condition. La prise en charge holistique est essentielle pour optimiser le développement et le bien-être des patients.
Recherches et Avancées Récentes
Au cours des dernières années, la recherche sur le syndrome d’Apert a considérablement progressé. Des études récentes montrent que des interventions précoces, notamment des traitements chirurgicaux et des thérapies de soutien, peuvent améliorer de manière significative les résultats à long terme. Par exemple, des études ont démontré que les enfants ayant reçu une prise en charge multidisciplinaire dès la naissance présentent des résultats fonctionnels améliorés par rapport à ceux recevant moins d’interventions.
Des recherches sur les mutations génétiques spécifiques associées au syndrome d’Apert continuent d’éclairer notre compréhension de la maladie. De nouvelles techniques de séquençage génétique permettent d’identifier plus facilement les mutations responsables, ouvrant la voie à des options thérapeutiques potentielles et à des conseils génétiques pour les familles.
Enfin, des études sur la qualité de vie des personnes atteintes du syndrome d’Apert mettent en lumière l’importance du soutien psychosocial et des ressources communautaires. Le soutien émotionnel et les groupes de soutien peuvent jouer un rôle essentiel dans le développement d’une identité positive et d’une résilience face aux défis uniques que présente cette condition.







