
La thalassémie est un trouble héréditaire complexe du sang, impactant la capacité du corps à produire de l’hémoglobine et des globules rouges. Ce phénomène peut se traduire par une quantité insuffisante de globules rouges et d’hémoglobine, ainsi que par des globules rouges de taille anormale.
Les conséquences de la thalassémie varient considérablement, allant d’une forme légère à des complications sévères, pouvant même être fatales. On estime qu’environ 100 000 nouveau-nés présentent chaque année des formes graves de thalassémie, touchant principalement des populations d’ascendance méditerranéenne, sud-asiatique et africaine.
Symptômes
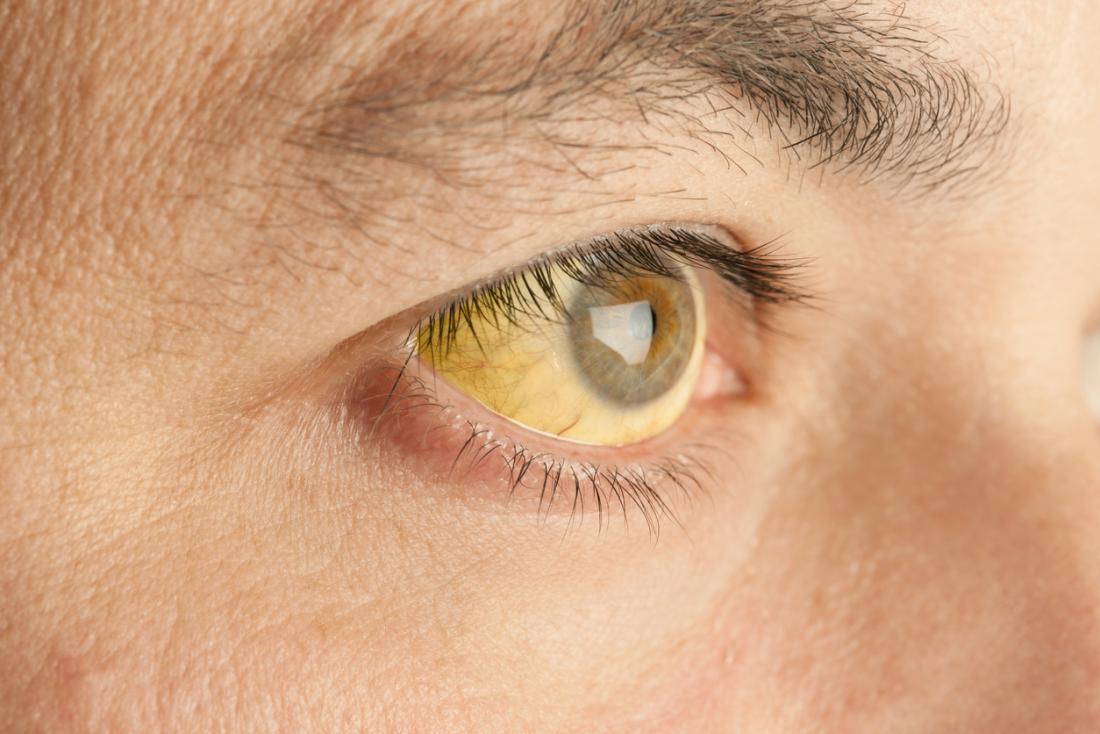
Les symptômes de la thalassémie peuvent grandement varier en fonction du type spécifique de la maladie. Chez la plupart des nourrissons atteints de bêta-thalassémie et de certains types d’alpha-thalassémie, les symptômes ne se manifestent généralement pas avant l’âge de 6 mois. Cela est dû à la présence d’hémoglobine fœtale, qui est remplacée par l’hémoglobine « normale » après cette période.
Les symptômes peuvent alors commencer à apparaître et incluent :
- jaunisse et teint pâle
- somnolence et fatigue persistante
- douleurs thoraciques
- mains et pieds froids
- essoufflement
- crampes dans les jambes
- palpitations cardiaques rapides
- appétit réduit
- retard de croissance
- maux de tête fréquents
- vertiges et évanouissements
- augmentation de la sensibilité aux infections
Des déformations squelettiques peuvent survenir lorsque le corps essaie de compenser par une production accrue de moelle osseuse. En cas de surcharge en fer, le corps tente d’absorber davantage de fer pour compenser, ce qui peut résulter de transfusions sanguines. L’excès de fer peut causer des dommages aux organes tels que la rate, le cœur et le foie.
Les individus atteints d’hémoglobine H sont particulièrement à risque de développer des calculs biliaires et une splénomégalie (hypertrophie de la rate). Si elles ne sont pas correctement gérées, les complications de la thalassémie peuvent entraîner une défaillance organique.
Traitement
Le traitement de la thalassémie dépend du type et de la gravité de la maladie. Pour les cas plus graves, des transfusions sanguines régulières peuvent être nécessaires pour maintenir des niveaux adéquats d’hémoglobine et de globules rouges.
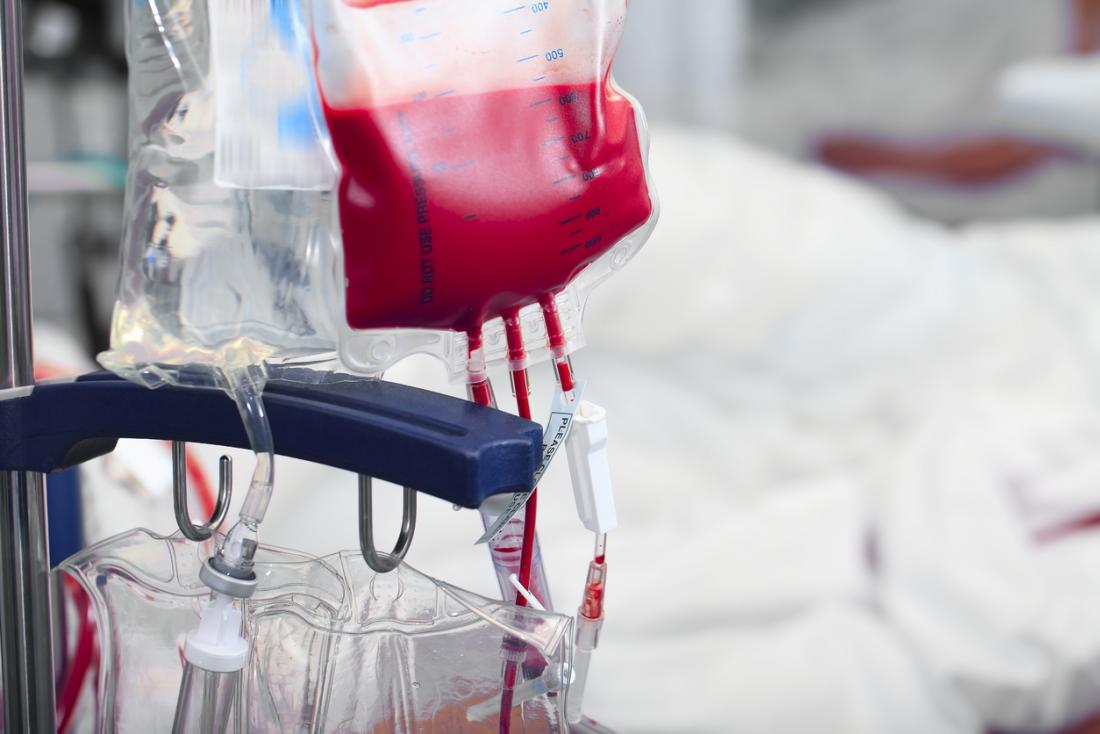
Les transfusions sanguines : elles permettent de reconstituer l’hémoglobine et les globules rouges. Les patients atteints de thalassémie majeure nécessitent entre huit et douze transfusions par an. Ceux ayant une forme moins sévère peuvent avoir besoin de jusqu’à huit transfusions par an, ou davantage en cas de stress, de maladie ou d’infection.
Chélation du fer : ce traitement vise à éliminer l’excès de fer dans le sang, souvent causé par des transfusions. Les médicaments comme la déféroxamine, administrée par injection, ou le déférasirox, pris par voie orale, sont couramment prescrits.
Les patients recevant des transfusions et une chélation peuvent également nécessiter des suppléments d’acide folique pour favoriser la production de globules rouges. En cas de thalassémie sévère, une greffe de moelle osseuse ou de cellules souches peut être envisagée, ainsi que des interventions chirurgicales pour corriger des anomalies osseuses.
La thérapie génique est un domaine de recherche prometteur, explorant des techniques pour traiter la thalassémie, comme l’insertion de gènes normaux ou la réactivation de gènes produisant l’hémoglobine fœtale.
Causes
L’hémoglobine, une protéine essentielle, transporte l’oxygène dans le corps via les globules rouges. La moelle osseuse utilise le fer provenant de notre alimentation pour produire de l’hémoglobine. Dans le cas de la thalassémie, la moelle osseuse ne parvient pas à produire suffisamment d’hémoglobine ou de globules rouges sains, entraînant une anémie et une fatigue.
Les formes légères de thalassémie peuvent ne pas nécessiter de traitement, alors que les cas plus graves requièrent des transfusions régulières.
Diagnostic

La majorité des enfants atteints de thalassémie modérée à sévère sont diagnostiqués vers l’âge de 2 ans. Les individus sans symptômes peuvent ne pas réaliser qu’ils sont porteurs de la maladie jusqu’à ce qu’ils aient un enfant affecté.
Différents tests sanguins permettent de diagnostiquer la thalassémie :
- Numération globulaire complète (CBC) : pour vérifier les niveaux d’hémoglobine et la taille des globules rouges.
- Numération des réticulocytes : elle évalue la rapidité de production des globules rouges par la moelle osseuse.
- Analyse du fer : pour déterminer si l’anémie résulte de la thalassémie ou d’une carence en fer.
- Tests génétiques : pour identifier la présence de la thalassémie ou de gènes défectueux.
- Tests prénataux : pour détecter la thalassémie chez le fœtus et évaluer la gravité potentielle.
Les types
L’hémoglobine est constituée de quatre chaînes de protéines alpha-globine et de deux chaînes de bêta-globine. Les deux principaux types de thalassémie sont l’alpha et la bêta.
Alpha thalassémie
Dans l’alpha-thalassémie, il y a une production insuffisante de protéines alpha. Cela est dû à des mutations sur les gènes alpha situés sur le chromosome 16. La gravité de la maladie dépend du nombre de gènes affectés.
Un gène défectueux : le patient ne présente aucun symptôme et est considéré comme porteur. Ce cas est connu sous le nom d’alpha-thalassémie minimale.
Deux gènes défectueux : le patient souffre d’une légère anémie, appelée alpha-thalassémie mineure.
Trois gènes défectueux : le patient développe une maladie de l’hémoglobine H, nécessitant des transfusions régulières.
Quatre gènes défectueux : l’alpha-thalassémie majeure est la forme la plus grave, souvent associée à l’hydrops fetalis, une condition menaçant la vie du fœtus.
L’alpha-thalassémie est plus fréquente en Asie du Sud-Est, en Chine, en Inde, au Moyen-Orient et en Afrique.
Bêta thalassémie
La bêta-thalassémie résulte d’une mutation sur l’un ou les deux gènes de globine nécessaires pour produire des chaînes de bêta-globine. La gravité de la maladie dépend du nombre de gènes mutés.
Un gène défectueux : il s’agit de la bêta-thalassémie mineure.
Deux gènes défectueux : cela peut entraîner des symptômes modérés à sévères, connu sous le nom de thalassémie majeure, anciennement appelée anémie de Colley.
La bêta-thalassémie est plus courante chez les personnes d’ascendance méditerranéenne, avec une prévalence élevée en Afrique du Nord, en Asie occidentale et aux Maldives.
Complications
Plusieurs complications peuvent découler de la thalassémie.
Surcharge de fer
Celle-ci peut résulter de transfusions sanguines fréquentes ou de la maladie elle-même, augmentant le risque d’hépatite, de fibrose, et de cirrhose.
Les dommages aux glandes endocrines, notamment à l’hypophyse, peuvent entraîner des retards de puberté et des troubles de croissance. Il existe également un risque accru de développer un diabète ou des troubles de la thyroïde.
La surcharge de fer peut également augmenter le risque d’arythmies et d’insuffisance cardiaque congestive.
Alloimmunisation
Il arrive qu’une transfusion sanguine provoque une réaction immunitaire, où le corps tente de détruire le sang transfusé. Une correspondance précise des groupes sanguins est cruciale pour éviter ce problème.
Rate agrandie
La rate, responsable du recyclage des globules rouges, peut devenir hypertrophiée, ce qui complique la gestion des cellules sanguines. Dans certains cas, une splénectomie peut être nécessaire, bien que cela entraîne des complications potentielles supplémentaires.
Infection
La splénectomie augmente le risque d’infection, tout comme les transfusions régulières qui exposent le patient à des maladies transmissibles par le sang.
Déformations osseuses
La moelle osseuse peut se développer, entraînant des déformations des os adjacents, notamment ceux du crâne et du visage, augmentant ainsi le risque de fractures.
Vivre avec la thalassémie
La gestion de la thalassémie exige des soins médicaux continus, en particulier pour ceux qui reçoivent des transfusions. Il est essentiel de respecter le calendrier des soins et de maintenir une attitude positive.

Les personnes atteintes de thalassémie sont encouragées à :
- participer à tous leurs rendez-vous médicaux réguliers
- maintenir un réseau de soutien et d’amis pour favoriser une attitude positive
- suivre une alimentation équilibrée pour préserver leur santé
- pratiquer une activité physique régulière
Certains aliments, comme les épinards et les céréales enrichies en fer, doivent être évités pour prévenir une surcharge en fer. Il est conseillé de discuter des choix alimentaires et d’exercice avec un professionnel de santé.
Les Centres de contrôle et de prévention des maladies (CDC) recommandent de tenir à jour les vaccinations pour prévenir d’éventuelles infections, en particulier chez ceux recevant des transfusions, qui sont plus exposés à des infections comme l’hépatite A ou B.
Thalassémie et grossesse
Les couples envisageant une grossesse devraient consulter un conseiller génétique, surtout s’ils ont des antécédents de thalassémie. Pendant la grossesse, les femmes atteintes de thalassémie peuvent faire face à des risques accrus de cardiomyopathie et de diabète, ainsi qu’à des restrictions de croissance fœtale.
Une évaluation par un cardiologue ou un hématologue est recommandée avant et durant la grossesse pour minimiser les risques, surtout pour celles atteintes de thalassémie bêta-mineure. Une surveillance fœtale continue peut aussi être nécessaire lors de l’accouchement.
Perspective
Les perspectives dépendent du type de thalassémie. Les personnes portant le trait thalassémique ont une espérance de vie normale. En revanche, les complications cardiaques associées à la bêta-thalassémie majeure peuvent réduire l’espérance de vie à moins de 30 ans.
Avancées récentes en recherche
En 2024, des recherches innovantes continuent d’émerger dans le domaine de la thalassémie. Une étude récente a mis en évidence l’importance des thérapies géniques, avec des résultats prometteurs permettant de corriger les défauts génétiques sous-jacents. Les essais cliniques pour de nouveaux médicaments ciblant la régulation de l’hémoglobine sont également en cours, offrant de nouvelles perspectives pour les patients.
De plus, un suivi régulier des niveaux de fer dans le sang grâce à des appareils portables pourrait révolutionner la gestion des patients, permettant des ajustements de traitement en temps réel. La prise de conscience et l’éducation autour de la thalassémie continuent de croître, contribuant à un meilleur soutien pour les patients et leurs familles.







